Method performance standards
Physico-chemical standards for biosimilars and other follow-on products: performance validation versus quality defining standards
All medicinal substances are required to meet stringent quality standards before they are approved for use in man, and biological medicines are no exception.
These quality standards include criteria of:
- identity – the substance is what it claims to be
- potency – it has the right strength
- safety – it is not going to cause adverse events such as toxicity or fever
- purity – it is free from contaminating substances which are not supposed to be there
Biological medicines are inherently the most complex substances used in man, and the battery of tests required to show that these quality standards are met is equally complex and extensive. They include both physico-chemical test methods, and specifically in the case of biological medicines, biological tests of activity.
Purity tests for biological medicines raise specific issues. Significant impurities in biological medicines are usually degraded forms of the medicinal product, for example dimer or polymers.
The presence of such dimerised or polymerised forms in a product would be of clinical significance as these are more likely to elicit antibodies to the product in man than the monomeric molecule.
This adverse reaction – immunogenicity – is an undesirable clinical consequence as it can both compromise the efficacy of the treatment or, in more severe cases, produce serious and irreversible pathology.
As a result, a usual element of the quality standard for a biological medicine is a test to show that it is free of dimers and higher polymers.
This test is most often a form of liquid chromatography known as size exclusion chromatography (SEC), and in validating the suitability of the test, the analyst would be required to prove that the test SEC method is both capable of separating dimer from monomer and of accurately determining their respective quantities.
For that a suitable reference standard would be needed which has defined levels of the dimer in it.
That requirement however exposes limitation in the suitability of current reference standards. The reference material for a therapeutic biological medicine is increasingly seen as being representative of the quality expected. In most cases this means that they should strive towards high purity.
The absence of specific impurities, such as the dimer, means that the reference material cannot be used to demonstrate system suitability.
This limitation of purified reference materials has been recognised for some time. Unlike synthetic chemicals, where impurities are usually by-products of the synthesis, most significant impurities of biological medicines are actually degradation products of the active substance such as dimers, aggregates, oxidised forms and cleaved forms. The approach to supporting these analytical methods has usually been to develop methods for generating the impurities from the purified reference substance such as heating, high pH and oxidation. These however are difficult to standardise and they produce products with limited utility. It is being increasingly recognised that the most suitable approach is to develop specific system suitability standards.
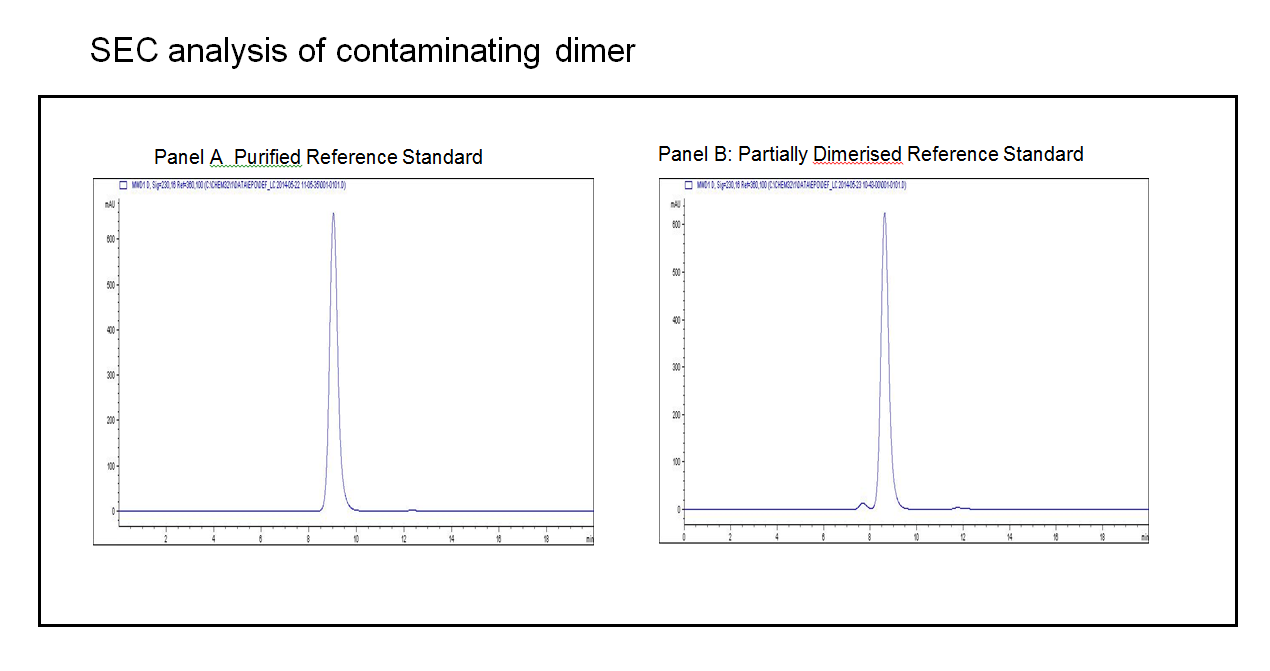
Panel A in the figure below illustrates the point. In this test the analytical target is the dimer, with a limit of 2%. To be suitable therefore the method must be able to clearly resolve the dimer (a small peak eluting just before the main peak), and quantify it at a level of 2%. The highly purified reference material for this substance (panel A) is essentially useless to support this function as it has no detectable dimer in it.
A further example of the utility of method performance standards is shown below, which considers a different analytic method known as peptide mapping.
In its traditional application, peptide mapping is an analytical test intended to address a different criterion of the quality standard – that of identity. Even simple biological medicines such as small proteins are structurally complex. A protein of molecular weight 20,000 Da, for instance, will have some 180 amino acids in its structure, and more than 2500 atoms, which is moreover folded into a globular structure.
The kind of analytical procedure available, such as chromatography, typically only address a limited number of features of this structure, and many possible structural changes would be undetected by such methods.
Proof of correct identity would therefore be difficult to infer. Peptide mapping addresses this limitation by specifically cleaving the molecule into a reproducible patter of smaller fragments and comparing the fingerprint obtained with that of a reference standard.
In a more advanced application, peptide mapping can be used as purity test, where it is considered capable of detecting contamination of a ‘correct’ protein, with smaller quantities of an ‘incorrect’ one. The figure below illustrates the ability of a peptide mapping procedure to resolve and quantify contamination of a recombinant protein with 10% of a variant where the glutamine at position 120 is substituted with histidine (Q120H).
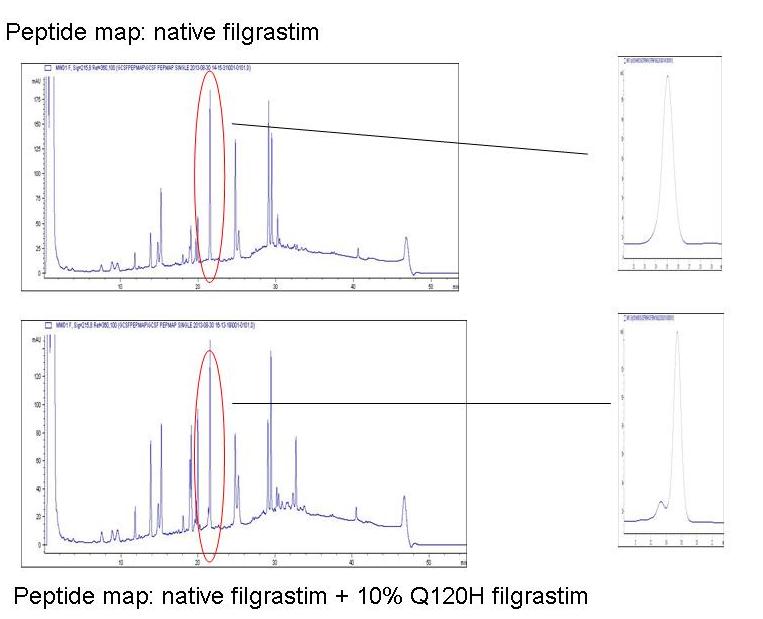 The fingerprints obtained with test and reference material are identical, other than the specific peak corresponding to the fragment with the potentially altered sequence, where the impurity shows up as a minor leading-edge shoulder.
The fingerprints obtained with test and reference material are identical, other than the specific peak corresponding to the fragment with the potentially altered sequence, where the impurity shows up as a minor leading-edge shoulder.
It can be readily appreciated that that an analyst would not be able to validate the performance of such a test method in detecting and quantifying specific sequence variant impurities without reference preparations of the targeted sequence variants.
Compliance with quality standards for Biological Medicines depends on a range of analytical methods including:
- various reverse-phase, ion-exchange and size exclusion High-performance liquid chromatography (HPLC) methods
- conventional gel and high performance capillary electrophoretic methods
- peptide mapping
- increasingly, spectroscopic methods including Nuclear Magnetic Resonance (NMR) and mass spectroscopy
All of these are to some extent dependent on reference materials, and the development of a range of reference materials to support these methods when they are used to detect and quantify specific impurities will be an increasing focus of activity at NIBSC.
The projects will be carried out in collaboration with key stakeholders such as Pharmacopoeias. They will supplement and develop the small successful programme of such standards that already exists, such as the molecular weight calibration standard for Low Molecular Mass Heparins and a system suitability standard for Filgrastim SEC recently developed in partnership with the United States Pharamacopoeia which is under evaluation.
As well as the examples shown above, such standards will also include oxidised, cleaved, reduced and deamidated proteins, as well as native and oxidised peptides to support peptide mapping applications.